当前位置:网站首页>单细胞代码解析-妇科癌症单细胞转录组及染色质可及性分析1
单细胞代码解析-妇科癌症单细胞转录组及染色质可及性分析1
2022-07-24 17:40:00 【小胡子刺猬】
这篇文章是2021年发表在MC上的文章,主要讲的是利用单细胞多组学的手段解析妇科癌症的文章。
文章题目:A multi-omic single-cell landscape of human gynecologic malignancies
doi:10.1016/j.molcel.2021.10.013
作者提供代码链接:https://github.com/RegnerM2015/scENDO_scOVAR_2020
作者说根据这个链接可以大致得把内容走一遍:ttps://github.com/RegnerM2015/scENDO_scOVAR_2020/wiki,因此我也是基本按照这个来进行代码解析的。
Introduction
这篇文章是为了解析妇科疾病的单细胞图谱。因此选用了手术切除后立即处理的11个人的卵巢和子宫内膜肿瘤的组织样本,进行单细胞的解离,构建了单细胞分辨率的转录组(SCRNA-SEQ)和染色质可及性(SCATAC-SEQ)图谱。这个稀有的数据集提供了解决这些肿瘤的复杂细胞异质性的内容,使研究人员能够将染色质可及性的变化与基因表达变化联系起来。同时这些数据提供了有关癌细胞如何重新利用和获取远端调节元件以驱动致癌转录模式发展。
以下为样本的来源:
作者还构建了相关的实验流程图:
cellranger and cellranger atac processing
在对单细胞进行解离后,获得下机数据,然后在cellranger上进行分析(因为作者选用的是10X的单细胞平台进行标记)。
转录组分析主要用的是CellRanger (version 3.1.0)。
##补充:参考基因组索引的构建
cellranger mkref --genome= GRCh38-3.0.0 \
--fasta=GRCh38-3.0.0.fa \
--genes=GRCh38-3.0.0.gtf \
--ref-version=3.0.0/Example_Patients1-2_scRNA-seq_mkfastq.sh
#!/usr/bin/env bash
#SBATCH --job-name HMGVCBGX9
#SBATCH -c 16
#SBATCH --mem 80g
#SBATCH --partition allnodes
#SBATCH --output HMGVCBGX9_demultiplex.job.out
#SBATCH --error HMGVCBGX9_demultiplex.job.err
##数据来源文件
DATA=/datastore/nextgenout5/share/labs/bioinformatics/seqware/francolab_10x_copy
##数据结果文件
OUT=/datastore/nextgenout5/share/labs/francolab/scRNA-seq_Endometrial.05.15.2019
##先对下机文件进行整合
cellranger mkfastq --id=HMGVCBGX9 \
--run=${DATA}/190514_NS500270_0297_AHMGVCBGX9 \
--csv=${OUT}/Samplesheet.csv \
--qc \
--localcores=16/Example_Patient1_scRNA-seq_CellRanger-count.sh
#!/usr/bin/env bash
##SBATCH:提交作业
#SBATCH --job-name 3533EL-RNA_F6
#SBATCH -c 16
#SBATCH --mem 80g
#SBATCH --partition allnodes
#SBATCH --output 3533EL-RNA_F6.cellranger-count.job.update.out
#SBATCH --error 3533EL-RNA_F6.cellranger-count.job.update.err
DATA=/datastore/nextgenout5/share/labs/francolab/Data
cellranger count --id=3533EL-RNA_F6_Update \
--fastqs=./fastq_path/HMGVCBGX9/3533EL-RNA_F6 \
--transcriptome=${DATA}/refdata-cellranger-GRCh38-3.0.0 \ ##参考基因组
--sample=3533EL-RNA_F6 \ ##根据不同的sample内容进行换名字
--localcores=16 \
--localmem=80ATAC分析主要用的是CellRanger ATAC (version 1.2.0)。
##参考基因组索引构建
##构建配置文件human.config
{
organism: "human" #物种明确后才能输出文件夹名称
genome: ["GRCh38-3.0.0"]
input_fasta: ["/path/to/reference/GRCh38-3.0.0.fa"]
input_gtf: ["/path/to/reference/GRCh38-3.0.0.gtf"]
non_nuclear_contigs: ["chrM"] #选做,移除线粒体部分注释信息
input_motifs: "/path/to/jaspar/motifs.pfm" #motif文件路径
}
##构建ATAC参考基因组
cellranger-atac mkref --config=/home/path/to/human.config/Example_Patients1-2_scATAC-seq_mkfastq.sh
#!/usr/bin/env bash
#SBATCH --job-name H333JBGXB_2
#SBATCH -c 16
#SBATCH --mem 80g
#SBATCH --partition allnodes
#SBATCH --output H333JBGXB_demultiplex.2.job.out
#SBATCH --error H333JBGXB_demultiplex.2.job.err
##数据来源
DATA=/datastore/nextgenout5/share/labs/bioinformatics/seqware/francolab_10x_copy
##数据结果文件
OUT=/datastore/nextgenout5/share/labs/francolab/scATAC-seq_Endometrial.05.16.2019
cellranger-atac mkfastq --id=H333JBGXB_2 \
--run=${DATA}/190515_NS500270_0298_AH333JBGXB \
--csv=${OUT}/Samplesheet.2.csv \
--qc \
--localcores=16/Example_Patient1_scATAC-seq_CellRanger-count.sh
#!/usr/bin/env bash
#SBATCH --job-name 3533EL-ATAC_A3
#SBATCH -c 16
#SBATCH --mem 80g
#SBATCH --partition allnodes
#SBATCH --output 3533EL-ATAC_A3.cellranger-count.job.update.out
#SBATCH --error 3533EL-ATAC_A3.cellranger-count.job.update.err
DATA=/datastore/nextgenout5/share/labs/francolab/Data
cellranger-atac count --id=3533EL-ATAC_A3_Update \
--fastqs=./fastq_path2/H333JBGXB/3533EL-ATAC_A3 \
--reference=${DATA}/refdata-cellranger-atac-GRCh38-1.2.0 \ ##参考基因组
--sample=3533EL-ATAC_A3 \
--localcores=16总结
在作者的关于本研究中使用的所提供数据格式的重要说明中提到:处理过的每个患者样本的scATAC-seq数据以片段文件的形式提供,或“类似于bed的表格文件,其中每一行表示检测所捕获的唯一ATAC-seq片段”。关于这种文件格式的更多信息,请访问Cell Ranger网站:https://support.10xgenomics.com/single-cell-atac/software/pipelines/latest/output/fragments。我们提供片段文件,而不是cellranger-atac生成的过滤过的峰条形码矩阵,因为我们使用片段文件作为在ArchR R包中执行的scATAC-seq分析的起始输入(Granja等人,2021年)。我们没有使用由cellranger-atac生成的过滤过的峰条形码矩阵,因为该算法以伪批量方式调用峰值(即使用来自样本中所有细胞的所有信号)。这种伪体积方法有效地掩盖了染色质可及性中的细胞类型特定模式,并将损害罕见细胞类型的ATAC信号的贡献(Granja等人,2021)。
作者提到的这个问题在我的分析中也遇到了这种情况,同时我们也采用这样的方法进行分析,也是比较有利于后续的分析的。
因此在分析单细胞数据的时候,首先是按照基本的流程进行分析,然后判断数据质量,然后对cellranger的软件的参数进行更改,是与以往的软件不太一样的,因此在第一步是需要进行大量调参的。
边栏推荐
- Shardingsphere database read / write separation
- Ipaylinks, a cross-border payment integration service, won the 3A Asia Award of treasury
- [how to optimize her] teach you how to locate unreasonable SQL? And optimize her~~~
- 2022 Asia International Internet of things exhibition
- Shardingsphere database read / write separation
- TCP协议调试工具TcpEngine V1.3.0使用教程
- ansible自动化运维详解(五)ansible中变量的设定使用、JINJA2模板的使用以及ansible的加密控制
- Array double pointer - deliberate practice
- CDN (content delivery network) content distribution network from entry to practice
- Logical operation of image pixels
猜你喜欢

Iftnews | Christie's launched its venture capital department, aiming at Web3 and metauniverse industries

2022 Asia International Internet of things exhibition

In the morning, Tencent took out 38K, which let me see the ceiling of the foundation

Getaverse,走向Web3的远方桥梁

NATBypass 端口转发

es(1)
How to remove the top picture of the bubble skin article details of solo blog

Df2net 3D model deployment
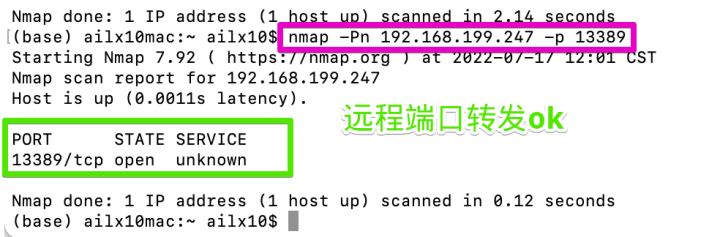
UFW port forwarding

Getaverse, a distant bridge to Web3
随机推荐
快速完成intelij idea的单元测试JUnit4设置
微信朋友圈的高性能复杂度分析
电脑监控是真的吗?4个实验一探究竟
Open source Invoicing system, 10 minutes to complete, it is recommended to collect!
How the computer accesses the Internet (IV) LAN and server response
Separation and merging of channels
JS image conversion Base64 Base64 conversion to file object
SV强制类型转换和常数
OpenCV 图片旋转
Tensorflow introductory tutorial (40) -- acunet
Class bytecode file
Memory allocation and recycling strategy
ufw 端口转发
C语言中的字符与字符串库函数的使用以及模拟实现
Colleges and universities have introduced 23 Philippine doctors with heavy funds, and the relevant departments have launched an investigation!
NC port forwarding
Review and analysis of noodle dishes
C语言自定义类型讲解 — 结构体
Niuke linked list solution record
Hcip day 3