当前位置:网站首页>Efficient integration of heterogeneous single cell transcriptome with scanorama
Efficient integration of heterogeneous single cell transcriptome with scanorama
2022-06-24 00:18:00 【tzc_ fly】

Abstract
Integration from multiple experiments 、 Laboratories and different technologies single-cell RNA sequencing(scRNA-seq) The data can reveal more abundant biological problems , But for now scRNA-seq Data integration methods are limited by the requirements of data sets from functionally similar cells . We proposed Scanorama Algorithm , The algorithm can identify and merge the shared cell types among all data set pairs , And accurately integrate scRNA-seq A heterogeneous collection of data . We apply Scanorama Consolidates and eliminates representatives from 9 Of different technologies 26 Different scRNA-seq Experimental 105,476 Batch effect of cells .Scanorama Sensitive to subtle temporal changes within the same cell lineage , Successfully integrated CD14+ monocyte (monocytes) They differentiate into macrophages at different stages of differentiation (macrophages) Cells with similar functions in time series data . Last , We show that Scanorama Several orders of magnitude faster than existing technology , You can make an appointment with 9 Consolidation within hours 1,095,538 Cells .
Main
independent single-cell RNA sequencing(scRNA-seq) Experiments have been used to discover new cell states and reconstruct cell differentiation trajectories . Through the efforts of global scientists , Researchers are currently generating large-scale 、 comprehensive scRNA-seq Data sets , These datasets describe a variety of cellular functions , It is expected to realize high-resolution observation of basic biology and disease processes . However , Due to the experimental batch 、 Differences in sample donors or experimental techniques , Combining large, unified reference datasets may be affected . Although recent methods have shown that , Can be integrated in multiple experiments scRNA-seq, But these methods automatically assume that all datasets share at least one common cell type , Or gene expression profiles share basically the same related structure in all data sets . therefore , These methods are prone to over correction , Especially in integrating scRNA-seq When there are very different data sets .
Here it is , We have put forward Scanorama: One way to effectively integrate multiple scRNA-seq Strategy for data sets , Even though they are composed of heterogeneous transcriptional phenotypes . Our method is similar to the computer vision algorithm for panoramic mosaic , The algorithm recognizes images with overlapping content , And merge these images into a larger panorama ( chart 1a). Again ,Scanorama Automatic identification of cells containing similar transcriptional profiles scRNA-seq Data sets , These matches can be used for batch correction and integration ( chart 1b).Scanorama Strong robustness to different data set sizes and sources , Data set specific content is preserved , And it is not required that all data sets share at least one cell type .
- chart 1:" panorama " Schematic diagram of data set composition .
- a: Panoramic mosaic algorithm finds and merges overlapping images , To create a larger composite image .
- b: A similar strategy can also be used to merge heterogeneous scRNA-seq Data sets .Scanorama Search for the nearest neighbor , To determine the shared cell type between all data set pairs . Based on hyperplane locally sensitive hash LSH And the dimension reduction technology of random projection tree and approximate nearest neighbor algorithm greatly accelerate the search speed . Linked cells form a matching relationship , It can be used to correct batch effects and combine them , Thus, the data set formed by connecting on the basis of these matches becomes scRNA-seq Of " panorama ".
Our method will match each other's nearest neighbors ( A technique for finding similar elements between two data sets ) Extend to find similar elements among multiple datasets . It was originally developed for pattern matching in images , Finding the nearest neighbor is also used to identify two at a time scRNA-seq Common cell types between datasets . However , To align more than two datasets , Existing methods select a data set as a reference , All other data sets are then integrated into the reference in turn , One at a time , This may lead to suboptimal results , And depends on the order in which the data set is considered . Even though Scanorama A similar approach is used when aligning sets of two data sets , But on larger data sets , It is not sequence sensitive , And it is not easy to make excessive correction , Because it can find a match between all data set pairs .
To optimize the process of searching for matching cells in all data sets , We introduced two key steps . We don't do the nearest neighbor search in the high-dimensional gene space , Instead, the gene expression matrix is used to perform effective random singular value decomposition for each cell (SVD), The gene expression profile of each cell was compressed into low dimensional embedding , This also helps to improve the robustness of the method to noise . Besides , We use approximate nearest neighbor search based on hyperplane locally sensitive hash and random projection tree , To greatly reduce the asymptotic and actual nearest neighbor query time .
Scanorama Can achieve scRNA-seq Data set integration and batch calibration . Even though Scanorama It will bring more computing costs , But it makes batch correction feasible for large data sets , Thus, more extensive downstream analysis can be carried out . for example , We can perform differential expression analysis on batch corrected gene expression data .
result

- chart 2:Scanorama Correctly integrated a simple data set set , Other methods have failed .
- a: We will Scanorama Applied to integrate three data sets : One is completely Jurkat cells (n=3257 Cells )、 One is completely 293T cells (n=2885 Cells ) And a 50/50 Proportionally mixed Jurkat and 293T Cell data set (n=3388 Cells ).
- b: Our method correctly puts Jurkat cells ( Orange ) and 293T cells ( Blue ) Integrate into two independent clusters .
- c and d: The existing scRNA-seq The data set integration approach is sensitive to the order in which it considers the data sets , And it's possible that Jurkat Data set and 293T Datasets are incorrectly merged , Forming clusters that do not correspond to the actual cell type :scran MNN The results of the integration are shown in c, and Seurat CCA The results of the integration are shown in d.

- chart 3: Across nine different sequencing technologies 26 Panoramic integration of single cell data sets .
- a: Use our method to 105476 Cells after batch correction t-SNE Distribution .
- b and c: other scRNA-seq Data set integration approach (scran MNN and Seurat CCA) Not designed for heterogeneous data set integration , Therefore, there is always a naive tendency to merge all data sets into a large cluster .
- d and e:Scanorama In less than 6 In minutes , Below 12 GB Of RAM in , Integrated 26 Of data sets 105476 Cells , This is better than the current scRNA-seq The integration approach is much more efficient .
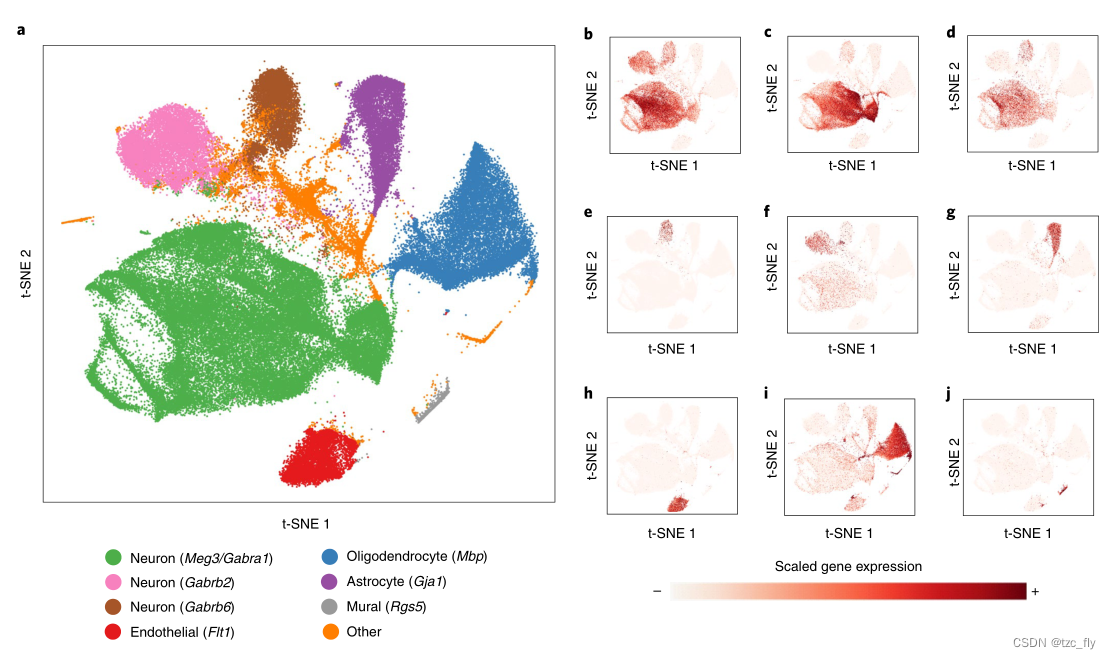
- chart 4:Scanorama Extensible to include 100 Integration of data sets of more than ten thousand cells .
- a:Scanorama It integrates the brain and spinal cord of mice 1095538 Cells .
- b To j: Use marker genes to reveal cell type specific clusters .

- chart 5:Scanorama Sensitive to subtle transcriptional changes in cellular state over time .
- a To c: The rows and columns of the heat map correspond to different data sets in the study of time processes ( Include data sets at the same point in time ). Higher comparison scores ( Navy Blue ) Tends to approach the diagonal , This indicates that the transcriptional similarity between datasets from more recent time points is greater . In every time series experiment , Time differences and alignment scores were significantly correlated .
- d To f: according to Monocle 2 The pseudo time allocated by the algorithm is visualized ,Scanorama Eliminates the CD14+ Batch effect of monocytes . But in the use of scran MNN After correction ,Monocle 2 The correct track can no longer be recognized . The original data are shown in d、Scanorama see e and scran MNN see f.
summary
The method proposed in this article is to integrate scRNA-seq Efficient solutions , Past ingest Different , There is no need to specify a specific reference data set , In fact, the idea of the past method is to integrate data into a larger cluster ( The domain is adapted to the reference data set ).Scanorama Using the idea of panoramic generation , It can realize the integration of multiple data sets at the same time , It can automatically match the same cell type under different data sets , At the same time, the differences of different cell types under different data sets are preserved , Is a more reasonable integration method .
Heterogeneity refers to the integration of information that retains the real differences between the two data sets .Scanorama Belongs to the realization of the correct elimination of technology , The batch effect brought about by the experiment .
边栏推荐
- Empty encoded password警告原因
- 如何利用數倉創建時序錶
- Learn PWN from CTF wiki - ret2text
- Comment utiliser l'entrepôt de données pour créer une table de synchronisation
- 9次Android面试经验总结,已收字节,阿里,高级android面试答案
- SQL database: summary of knowledge points, no suspension at the end of the period
- [new cos series] | object storage cos data Vientiane CI processing special data processing resource package 4.5 folded into a big gift package!
- Easycvr implementation of adding preset point position function of Dahua equipment
- 解决项目依赖报红问题
- Android App bundle exploration, client development interview questions
猜你喜欢

Expander+listbox of WPF effect
kubernetes之常用核心资源对象

NLP工程师是干什么的?工作内容是什么?

【面试经验包】面试被吊打经验总结(一)

Revit API: schedule viewschedule

Learn PWN from CTF wiki - ret2text

What are the two types of digital factories
What should I pay attention to in the interview of artificial intelligence technology?

抖音实战~手机号密码一键注册登录流程(限制手机终端登录)

合成大西瓜小游戏微信小程序源码/微信游戏小程序源码
随机推荐
Salesforce batch apex batch processing (V) asyncapexjob intelligence
如何入门机器学习?
Keywords such as extern and struct
抖音实战~密码找回
Complete collection of development environment configuration -- Visual Studio 2022 installation
C语言:关于矩阵右移问题
规律/原理/规则/法则/定理/公理/本质/定律
社招面试必不可少——《1000 道互联网大厂 Android工程师面试题》
Android 72021 latest Android interview written test questions sharing
Android App Bundle探索,客户端开发面试题目
SAVE: 软件分析验证和测试平台
Jimureport building block report - table linkage chart topic
抓取开机logcat
Hackers can gain administrator privileges by invading Microsoft mail server and windows Zero Day vulnerability November 23 global network security hotspot
解决base64压缩文件,经过post请求解压出来是空格的问题
【osg】OSG开发(04)—创建多个场景视图
Android Aidl: cross process call service (Aidl service), kotlininvoke function
国内首款开源MySQL HTAP数据库即将发布,三大看点提前告知 石原子科技重磅推出
Leetcode - linked list written test questions
Fix blog theme php8 compatibility and optimize dark mode